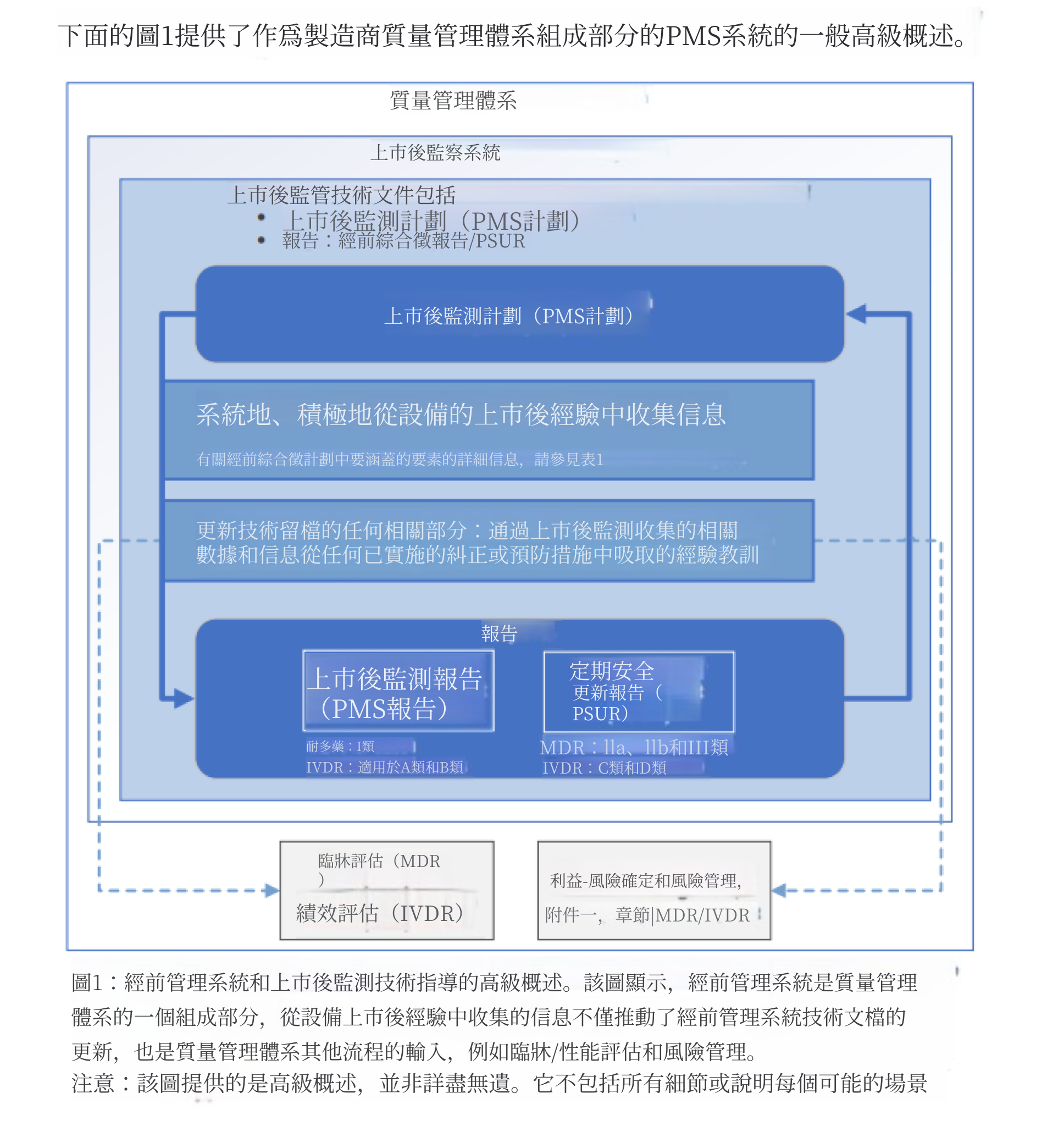
MDR 與 IVDR 架構下之上市後監督(PMS):法規脈絡與制度架構
QMS、PMS 系統、技術文件(含 PMS 計劃及 PMS 報告/PSUR)
於歐盟《醫療器材法規》(Regulation (EU) 2017/745, MDR)及《體外診斷醫療器材法規》(Regulation (EU) 2017/746, IVDR)之制度架構下,上市後監督(PMS)被界定為一項具結構性、前瞻性且持續運作之制度,並構成製造商品質管理體系(QMS)的組成部分。根據 MDR 第 10(10) 條和第 83 條 / IVDR 第 78 條的規定,PMS 系統旨在主動、系統地收集、記錄和分析設備在整個生命週期中的品質、安全性和性能數據。
該系統建立在 PMS 計劃之上(MDR 第 84 條 / IVDR 第 79 條,詳見兩部法規的附件 III),該計劃定義了將收集哪些資訊、資訊來源以及收集方法。上市後監督的技術文件是整體技術文件的一個明確子集,主要包括 PMS 計劃,以及 PMS 之產出文件,即 PMS 報告和/或定期安全更新報告(PSUR),具其適用取決於醫療器材之分類及相應之法規架構。該系統的輸出透過 PMS 報告(MDR 第 85 條 / IVDR 第 80 條,適用於 I 類器械和 A、B 類 IVD)或定期安全更新報告(PSUR)(MDR 第 86 條 / IVDR 第 81 條,適用於 IIa、IIb 和 III 類器械以及 C、D 類 IVD)形式化呈現。
正如 MDCG 2025-10 的高層概覽所示,PMS 計劃推動了上市後經驗的持續收集,這些經驗反過來被納入技術文件,並直接與其他關鍵 QMS 流程(如風險管理和效益–風險評估〔MDR/IVDR 附件 I〕以及臨床或性能評價)相互銜接,確保真實世界數據系統性地支持監管合規和生命週期決策。
核心資訊
PMS 必須制度必須具備結構化、前瞻主動並涵蓋產品全生命週期之特性。
MDCG 2025-10 重申了 MDR/IVDR 的核心原則:PMS 並非僅屬於被動的投訴處理活動,而是一項持續性、具結構性且具前瞻主動性的制度。該制度應貫穿醫療器材之整個生命週期,自產品首次上市或投入使用起,直至市場上最後一件產品之預期使用壽命終止為止。此外,文件亦強調,上市後監督之規劃應於醫療器材開發階段即開始進行,以便事先界定在產品上市後將採取哪些監測與資料蒐集活動,從而系統性地蒐集與評估醫療器材實際使用情境下之性能相關資訊。
兩個重點值得關注:
- 「前瞻主動(Proactive)」並非可選性用語。指引明確指出,前瞻主動式的PMS 是有意地尋求超越投訴管道的資訊,例如文獻篩查、使用者回饋、註冊/登記(在適用情況下)、客戶調查、PMCF/PMPF 等。
- PMS 輸出必須驅動決策。 結論及後續行動需要透過 PMS 報告或 PSUR 加以記錄與文件化。下一循環之 PMS 活動亦可能需要修訂既有之 PMS 計畫,以確保上市後監督制度能持續反映最新之市場與使用資訊。
MDCG 2025-10 的實用價值
可與品質管理系統( QMS) 對應整合的「PMS 運行模型」
指引將 PMS 描述為一個實際的循環:
資訊來源 → 收集 → 評估/分析 → 結論 → 行動措施 →品質管理系統(QMS)與技術文件之更新
這不僅是概念性的。文件強調 PMS 資訊會持續用於更新關鍵的 QMS 要素,尤其是風險管理、效益–風險評估以及臨床/性能評價。
換句話說,稽核員可能會提出類似的問題: 「請說明 PMS 信號如何觸發風險管理文件、臨床評價報告、標籤/說明書 (IFU) 以及 CAPA 決策的更新。」
「前瞻主動式資料蒐集」之要求進一步明確化
(對 PMS 計劃之要求門檻有所提升)
MDCG 2025-10 強調 PMS 計劃是整個系統的引擎。它重申計劃必須定義監測內容、頻率以及方法,並根據風險等級、器械類型和真實使用場景來選擇。表 1 特別有用,簡明總結了 PMS 計劃必須涵蓋的要素(附件 III 第 1 節)。
審計員可能會關注的計劃要素包括:
- 明確界定之指標與門檻值:用以支援對效益–風險平衡及風險管理之持續再評估,而非僅止於「追蹤客訴趨勢」等概括性描述。
- 趨勢通報之準備機制:包括用以偵測事件發生頻率或嚴重程度出現統計上顯著增加之方法或程序,以及所採用之觀察期間。
- 客訴調查方法:應採用與產品風險程度相稱之有效工具與方法,尤其對於較高風險等級之產品組合更為重要。
- 可比/「類似產品」之資訊蒐集與分析:作為系統化的輸入,結合最新技術(SOTA)監測,而不是非正式的市場傳聞。
一個細微但重要的點:指南建議 PMS 計劃可以定義「使用哪些方法」,而具體的「如何/由誰執行」可以放在引用的 SOP 中,只要計劃保持具體且可追溯。
數據品質的重要性
指引明確警告須留意資訊來源之可靠性。它特別提到不可驗證的數據(甚至包括公共/社交媒體)可能導致過度反應,並提醒製造商在分析前必須考慮數據品質與完整性。
此一要求並不意味著應完全忽略此類「噪音較多」之資訊來源;然而,製造商應在文件中清楚說明以下事項:
- 如何對此類資訊進行初步篩選與分級處理
- 如何進一步進行佐證或交叉驗證,或在何種情況下決定不予採信
- 如何避免此類資料對趨勢分析或矯正與預防措施(CAPA)啟動決策產生不當影響
客製化醫療器械 (CMDs):PMS 要求被明確強化
MDCG 2025-10 專門說明定制醫療器械並不豁免於 MDR 的 PMS 要求。製造商仍需建立 PMS 系統,並規劃/記錄生產後的經驗,包括 PMCF,並透過分組(相同用途/材料/工藝/設計原則)來管理,而不是將每個 CMD 獨立作為完整生命週期文件。
CMD 製造商必須:
- 第一類客製化醫療器材:應編製上市後監督報告
- 第二類 a、第二類 b 及第三類客製化醫療器材:應編製定期安全更新報告(PSUR)
製造商的重點行動方向
若製造商希望快速且具可辯護性地與 MDCG 2025-10 保持一致,以下幾項措施通常能帶來最高的實務效益:
1. 壓力測試 PMS 計劃是否符合附件 III 要求
- 前瞻主動之資訊來源
- 資料分析方法
- 監測指標與門檻值
- 客訴調查方法
- 趨勢分析與趨勢通報方法,以及所採用之觀察期間
- 通報與溝通程序,包括主管機關、公告機構、經濟營運者及使用者等相關利害關係人
- 用以界定矯正措施範圍之可追溯性工具
2. 證明 PMS 與風險管理及評估活動之回饋循環
指引文件明確指出:PMS 發現必須持續回饋到效益–風險與風險管理,以及臨床/性能評價。如果 PMS 識別出新的副作用或產品缺失,風險管理流程必須跟進。 一個實用的證據方式是維護簡單的「信號到更新」追蹤: PMS 信號 → 評估記錄 → 決策(無需行動 / 啟動 CAPA / 啟動 FSCA / 標示或使用說明書修訂 / 臨床評估報告更新) → 更新文件引用。
3. 在 PMS 報告/PSUR 中清晰呈現「結論 + 行動」
文件強調結論和後續行動必須記錄在 PMS 報告或 PSUR 中,並且 PMS 計劃可能需要根據循環結果進行修訂。稽核員尤其喜歡看到這個閉環。
這並非新法規,但將形塑監管期望
MDCG 指南並不像法規那樣具有法律約束力(文件本身也包含標準免責聲明),但它強烈影響主管當局和公告機構對「良好實踐」的解讀。
因此,即使你已經在執行 PMS,問題也變成:你的 PMS 系統是否符合 MDCG 2025-10 所描述的主動、基於風險、與 QMS 深度整合的模型?