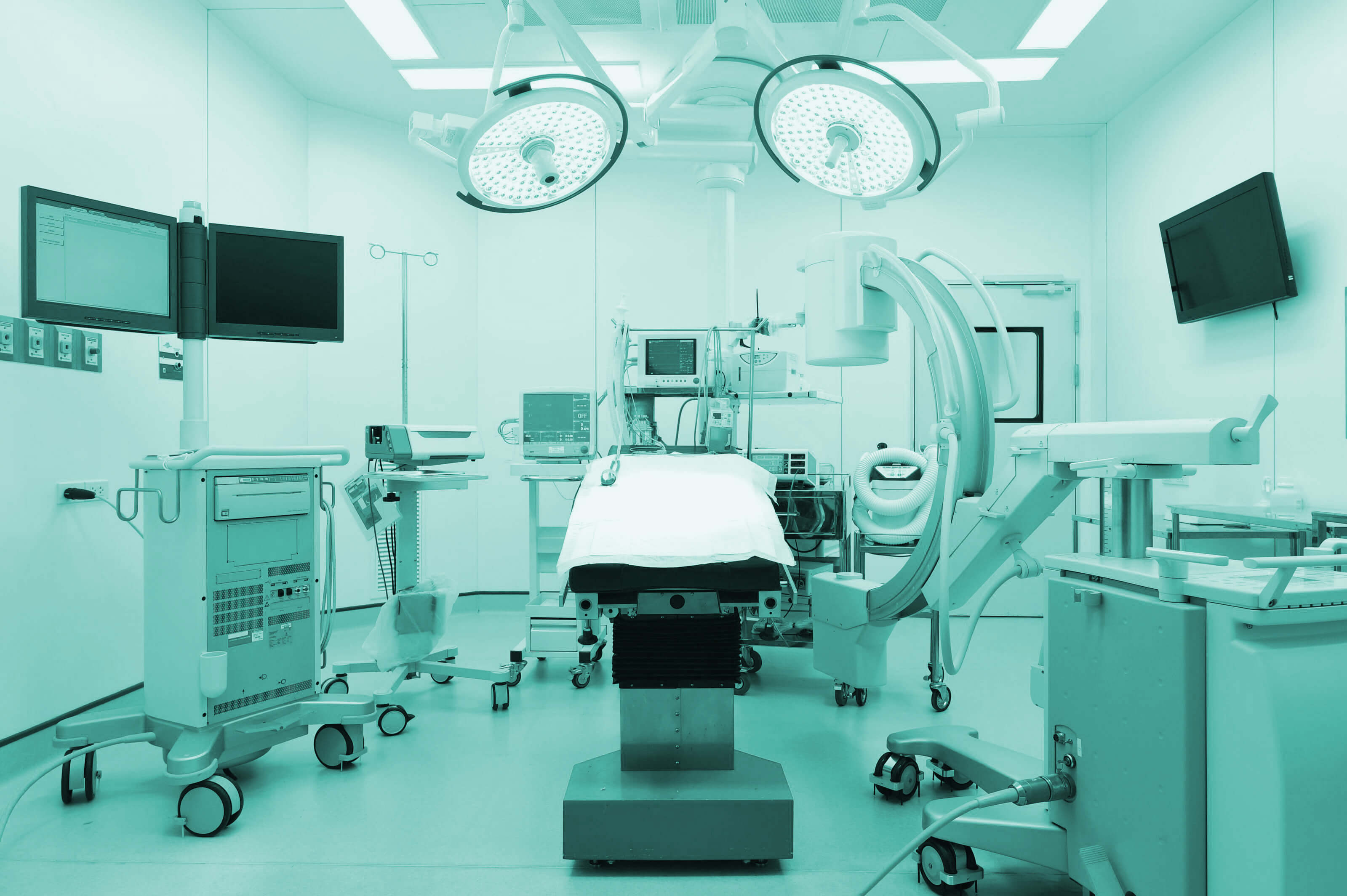
Одобряване на медицински изделия за продажба или разпространение в Европейското икономическо пространство
Правно изискване в икономическото пространство на ЕС
Подчертаване на компетентността на Вашата компания
Демонстриране на съответствие с най-високите продуктови стандарти
Маркировката СЕ осигурява достъп до допълнителни пазари

Loading...
Нов Регламент (ЕС) 2017/745 за медицинските изделия, приет от Европейския парламент

Loading...
Съществени промени за производителите на медицински изделия

Loading...
Промените в Регламент (ЕС) 2017/745 за производителите на медицински изделия

Loading...
Промените в Регламент (ЕС) 2017/745 за нотифицираните органи

Loading...
Какво може да направи за Вас DQS Medizinprodukte GmbH като нотифициран орган

Loading...
Процес на сертификация съгласно MDR (EU) 2017/745
За да можем да Ви предложим нашите услуги, първо се нуждаем от описание на планирания от Вас проект за сертифициране, както и от известна информация, свързана с продукта. От особена важност са планираната от Вас процедура за съответствие, както и предназначението и класификацията на риска на Вашите медицински изделия.
Моля да ни предоставите тази информация в предвидения за тази цел формуляр, т.нар. Основни данни.
След като проверим подадената от Вас информация, ще Ви изпратим предварителна оферта, включително цената за одита на СУК и оценката на Вашата техническа документация.
Заедно с офертата ще получите и формулярите за кандидатстване.
За да приемете нашата оферта за сертификация, моля да изпратите попълнения формуляр за кандидатстване.
Важна забележка: Както е посочено във формуляра за кандидатстване, Вашата процедура за оценяване на съответствието съгласно Регламент (ЕС) 2017/745 започва с получаването на попълнения формуляр. Самото заявление не е гаранция за сертифициране. Моля, обърнете внимание на нашите задължения за докладване, които са изложени в нашите Общи условия (ОУ).
След като получим Вашия формуляр за кандидатстване, ние ще прегледаме и оценим данните и информацията, които сте подали. За изясняване може да се наложи да поискаме от Вас да предоставите допълнителна информация и подробности.
При успешно приключване на прегледа, Вашето заявление за сертификация ще бъде прието. Ако на този етап бъдем принудени да отхвърлим Вашето заявление по технически или формални причини, това ще доведе до задължения за уведомяване от наша страна, както е описано в нашите Общи условия.
На първия етап се прави необходимата оценка на Вашата техническа документация на базата на извадков план. За тази цел от Вас ще бъде поискано да представите всички необходими документи за оценка. Ще получите резултата от всяка оценка под формата на окончателен доклад след вземането на решение за сертификация (вж. следващата стъпка).
Втората стъпка е системната оценка на Вашата система за управление на качеството. Тя се извършва на етапи, които включват анализ на системата (етап 1) на базата на документацията на вашата СУК и оценка на системата на място (етап 2). Резултатите от оценката на системата също ще Ви бъдат изпратени в отделни доклади, веднага след като бъдат взети решенията за сертификация в следващия етап.
Моля обърнете внимание, че в хода на оценките могат да възникнат изисквания за докладване или дори прекратяване на процедурите за оценка. Възможните действия, както и съответните последствия са описани в нашите Общи условия.
Резултатите от оценяването на Вашата техническа документация, както и оценката на Вашата система, се проверяват от независима група експерти, които потвърждават или отхвърлят препоръката за сертификация на оценителя. Ако има някакви въпроси относно съдържанието, ние ще се свържем с Вас.
Самото решение за сертификация е многоетапен процес с няколко стъпки за вътрешен контрол, за да се гарантира че решенията за сертификация са правилни, и че са предприети подходящи действия.
За да поддържаме Вашата сертификация, трябва да извършваме надзорни дейности на редовни интервали от време. Те се състоят от ежегодни одити на Вашата система за управление на качеството, оценки на Вашата техническа документация в съответствие с определен извадков план, както и необявени одити.
Ако в рамките на един сертификационен цикъл настъпят промени във Вашата система за управление на качеството или в техническата документация, необходимите надзорни дейности от наша страна ще бъдат включени в одитния цикъл.
Сертификацията съгласно Регламент (ЕС) 2017/745 е с максимална продължителност пет години и може да бъде продължена с последващ одитен цикъл след подаване на заявление за ресертификация.

Loading...
Цена на сертификацията в съответствие с Регламент (ЕС) 2017/45

Loading...
Защо да изберете DQS MED за сертификация по MDR?
- Широкообхватно продуктово портфолио от разнообразни програми за нормативна и регулаторна сертификация от една сертификационна организация
- Световна мрежа с над 200 опитни оценители и експерти
Вижте повече
Вижте по-малко
Basic data on the Medical Devices Regulation MDR (EU) 2017
If you are interested in a certification of a medical device according to MDR, you are at the right place.
We are pleased that you are interested in our certification and assessment services. We will be happy to provide you with a non-binding cost estimate free of charge. For this we need some information about your company. Please fill out the basic MDR data and send it us. Please note that we also need the basic data list MP MDR for the procedure.
